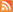
Key words :
evolution,
organism
,treponematoses
,genetics
,phylogenetic tree
,évolution
,syphilis
,variation
,colonization
Syphilis Origins Solved?
21 Jan, 2008 12:09 pm




A new paper in PLoS Neglected Tropical Diseases attempts to tackle a rather sensitive topic...
There was an extensive article about this study in the New York Times on 1/15/08 (by John Noble Wilford). Overall the Times article is good (except Wilford gets the definition of phylogenetic analysis a bit wrong - saying it is the study of the evolutionary relationships between organisms when really it is the study of evolutionary relationships of anything... but hey that is OK).
I confess I am not sure if I am completely convinced by all of Harper et al's arguments concerning the evolution of these bugs. My main concern is that the amount of variation they observe (in ~ 20 genes across these strains) is very very low. And thus the resolution of the phylogenetic trees is quite poor.
Because this is a PLoS paper, it is truly "Open Access" and I can include the Figure here in my blog as long as I cite the original source (see below). If you look at the tree you can see some #s on the branches in the tree. These are based on a statistical test called bootstrapping and the numbers indicate (roughly) how well the tree that is shown represents all of the polymorphisms in the data. The #s are percentages and alas the % support is not very high for many of the branches. So a better resolution of the question of the origin of these diseases will likely require, well, a better resolution on the tree. This in turn will likely require complete genome sequences and perhaps more strains samples. Nevertheless, given the results they have, their arguments seem sound ... and this should stimulate people to gather more genomic data from these bugs.
Also see some other blogs on this
- Neil Woodburn
- John Dennehy (who mentions an article by Carl Zimmer in a non OA publication ... find out which by going to his blog)
Here is Figure 3. It is from Harper KN, Ocampo PS, Steiner BM, George RW, Silverman MS, et al. (2008) On the Origin of the Treponematoses: A Phylogenetic Approach. PLoS Negl Trop Dis 2(1): e148. doi:10.1371/journal.pntd.0000148
Figure 3. This maximum likelihood tree is based on 20 polymorphic regions in the T. pallidum genome. Bootstrap support was estimated with 1,000 replicates in order to assess confidence at branching points and are shown within circles where values are high (>90%). Bootstrap support values for both maximum likelihood and maximum parsimony trees are shown, in that order.
Originally published on: The Tree of Life
Key words :
